
Pessoal, tenho uma pergunta:
Existe uma maneira de obter 3,4-Metilenodioxifenilpropan-2-ona (MDP2P) a partir do Eugenol?
Se sim, alguém poderia me dar uma síntese precisa?
Encontrei essa síntese, mas não entendo como posso produzir beta-nitroisoeugenol e β-Nitroisosafrole.
O problema é que em meu país é impossível obter óleo de sassafrás, safrol, isosafrol e piperonal.
Em um balão de 5 litros com 3 gargalos, equipado com um agitador e um condensador de refluxo, foram colocados 1.000 mL de etanol e 110 g de beta-nitroisoeugenol. A mistura foi aquecida com agitação e, quando o nitroisoeugenol foi dissolvido, foram adicionados 2500 mL de água quente. Com aquecimento e agitação vigorosa, foram adicionados 200 g de pó de ferro reduzido e 8 g de cloreto férrico hidratado. Com agitação contínua, 100 ml de ácido clorídrico concentrado foram adicionados lentamente. O ácido clorídrico causou uma reação violenta que diminuiu após cerca de 5 minutos; a mistura foi refluxada com agitação por duas horas e, em seguida, destilada sob pressão reduzida até que aproximadamente 2 litros de destilado fossem coletados. O resíduo foi filtrado e o óxido de ferro macio foi lavado cuidadosamente com água quente e depois com éter. O filtrado combinado e as lavagens foram fortemente acidificados com ácido clorídrico e extraídos com éter. O éter foi seco e destilado para produzir um óleo claro que foi fracionado no vácuo para dar 68 g de vanililmetilcetona com ebulição de 126-127°C a 0,3 mm (72% de rendimento).
A 3,4-dimetoxifenilacetona e a 3,4-metilenodioxifenilacetona foram preparadas a partir dos β-nitropropenilbenzenos correspondentes por meio de procedimentos quase idênticos com 90 e 72% de rendimento.
Para a preparação de 2-MeO-P2P com tolueno/água como solvente, consulte Organic Syntheses, Coll. vol. 4, p. 573.
Minha modificação dessa preparação:
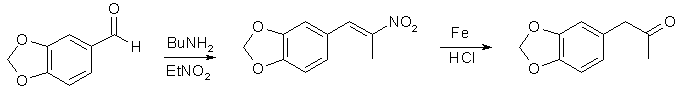
Em um balão de 2 litros com 3 gargalos, equipado com um agitador e um condensador de refluxo, foram colocados 460 ml de etanol1 e 67 g de β-nitroisosafrol2. A mistura foi aquecida com agitação e, quando os cristais amarelos foram dissolvidos, foram adicionados 1100 ml de água quente. Com aquecimento e agitação vigorosa, foram adicionados 80 g de pó de ferro reduzido3 e 5 g de FeCl3-6H2O. Com agitação contínua, 63 ml de HCl conc. foram adicionados em 30 minutos. A mistura foi refluxada com agitação por 2 h e, em seguida, destilada sob pressão atmosférica4. O resíduo foi filtrado e o óxido de ferro marrom foi extraído 3x50 ml de CH2Cl2. O filtrado e as lavagens orgânicas foram acidificados com HCl e a camada de cetona vermelha foi coletada. A camada de água verde-clara foi extraída 2x100 ml de CH2Cl2. Os extratos combinados foram secos com Na2SO4 e destilados à pressão atmosférica (banho a 80°C). O diclorometano foi completamente removido no vácuo para dar 55 g de MDP2P bruto como óleo vermelho escuro, que foi usado para a reação CH3NH2/Al.
Observações:
Existe uma maneira de obter 3,4-Metilenodioxifenilpropan-2-ona (MDP2P) a partir do Eugenol?
Se sim, alguém poderia me dar uma síntese precisa?
Encontrei essa síntese, mas não entendo como posso produzir beta-nitroisoeugenol e β-Nitroisosafrole.
O problema é que em meu país é impossível obter óleo de sassafrás, safrol, isosafrol e piperonal.
Em um balão de 5 litros com 3 gargalos, equipado com um agitador e um condensador de refluxo, foram colocados 1.000 mL de etanol e 110 g de beta-nitroisoeugenol. A mistura foi aquecida com agitação e, quando o nitroisoeugenol foi dissolvido, foram adicionados 2500 mL de água quente. Com aquecimento e agitação vigorosa, foram adicionados 200 g de pó de ferro reduzido e 8 g de cloreto férrico hidratado. Com agitação contínua, 100 ml de ácido clorídrico concentrado foram adicionados lentamente. O ácido clorídrico causou uma reação violenta que diminuiu após cerca de 5 minutos; a mistura foi refluxada com agitação por duas horas e, em seguida, destilada sob pressão reduzida até que aproximadamente 2 litros de destilado fossem coletados. O resíduo foi filtrado e o óxido de ferro macio foi lavado cuidadosamente com água quente e depois com éter. O filtrado combinado e as lavagens foram fortemente acidificados com ácido clorídrico e extraídos com éter. O éter foi seco e destilado para produzir um óleo claro que foi fracionado no vácuo para dar 68 g de vanililmetilcetona com ebulição de 126-127°C a 0,3 mm (72% de rendimento).
A 3,4-dimetoxifenilacetona e a 3,4-metilenodioxifenilacetona foram preparadas a partir dos β-nitropropenilbenzenos correspondentes por meio de procedimentos quase idênticos com 90 e 72% de rendimento.
Para a preparação de 2-MeO-P2P com tolueno/água como solvente, consulte Organic Syntheses, Coll. vol. 4, p. 573.
Minha modificação dessa preparação:
Em um balão de 2 litros com 3 gargalos, equipado com um agitador e um condensador de refluxo, foram colocados 460 ml de etanol1 e 67 g de β-nitroisosafrol2. A mistura foi aquecida com agitação e, quando os cristais amarelos foram dissolvidos, foram adicionados 1100 ml de água quente. Com aquecimento e agitação vigorosa, foram adicionados 80 g de pó de ferro reduzido3 e 5 g de FeCl3-6H2O. Com agitação contínua, 63 ml de HCl conc. foram adicionados em 30 minutos. A mistura foi refluxada com agitação por 2 h e, em seguida, destilada sob pressão atmosférica4. O resíduo foi filtrado e o óxido de ferro marrom foi extraído 3x50 ml de CH2Cl2. O filtrado e as lavagens orgânicas foram acidificados com HCl e a camada de cetona vermelha foi coletada. A camada de água verde-clara foi extraída 2x100 ml de CH2Cl2. Os extratos combinados foram secos com Na2SO4 e destilados à pressão atmosférica (banho a 80°C). O diclorometano foi completamente removido no vácuo para dar 55 g de MDP2P bruto como óleo vermelho escuro, que foi usado para a reação CH3NH2/Al.
Observações:
- O uso de metanol em vez de etanol levou à dissolução incompleta dos cristais amarelos.
- O β-Nitroisosafrol pode ser convenientemente preparado por Knoevenagel: 150 g de piperonal foram misturados com 76 ml de nitroetano (o piperonal deve se dissolver completamente para dar um líquido incolor) e 1,5 ml de n-butilamina(ou n-pentilamina) foi adicionado. A solução amarela resultante foi mantida em um local escuro; um pouco de água começou a aparecer depois de 2 a 3 dias, conforme a reação prosseguia. Cristais laranja-claros do produto começaram a se precipitar após 6 a 8 dias. Após 20 dias, a mistura foi filtrada. O filtrado marrom claro foi filtrado novamente após 5 dias para obter a próxima safra do produto. Rendimento de 85-90% de cristais laranja claro, que são adequados para a preparação da cetona.
- Foi usado um ferro reduzido com hidrogênio. O uso de pó de ferro de 20 mesh em vez de ferro reduzido levou a uma reação menos vigorosa, o etanol resultante não continha NH3 e a cetona foi contaminada com cerca de 20% de sua oxima (redução não quantitativa).
- Uma fração de 77-85°C foi coletada. Ela continha algum NH3 que pode ser neutralizado com H2SO4 conc. e destilado novamente por meio da coluna Vigreux. O etanol 90% resultante foi usado para a preparação da próxima porção de cetona.
