
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 3,064
- Solutions
- 3
- Reaction score
- 3,501
- Points
- 113
- Deals
- 1
Extraction theory.
"Extraction" refers to transference of compound(s) from a solid or liquid into a different solvent or phase. When a tea bag is added to hot water, the compounds responsible for the flavor and color of tea are extracted from the grounds into the water. Decaffeinated coffee is made by using solvents or supercritical carbon dioxide to extract the caffeine out of coffee beans. In the chemistry lab, it is most common to use liquid-liquid extraction, a process that occurs in a separatory funnel. A solution containing dissolved components is placed in the funnel and an immiscible solvent is added, resulting in two layers that are shaken together. It is most common for one layer to be aqueous and the other an organic solvent. Components are "extracted" when they move from one layer to the other. The shape of the separatory funnel allows for efficient drainage and separation of the two layers.
Fig.1 Schematic of extraction

Compounds move from one liquid to another depending on their relative solubility in each liquid. A quick guide to solubility is the "like dissolves like" principle, meaning that non-polar compounds should be readily extracted into non-polar solvents (and vice versa). The compounds responsible for the taste and color of tea must be polar if they are readily extracted into hot water. When allowed to equilibrate between two liquids in a separatory funnel, the majority of a compound often ends up in the layer that it is more soluble.
Fig.2

Step-by-Step Procedures For Extractions.
Single Extraction.
The pictures in this section show a single extraction of methyl red (colored compound, Fig. 3) from an aqueous solution (bottom layer) into 25 ml of ethyl acetate (top layer). The aqueous solution originally has a pink color, as the methyl red appears red in acidic solution (the aqueous solution was made from 50 ml water, 5 drops of 0.1MHCl and 5 drops of 1% methyl red indicator solution). The methyl red has a large partition coefficient and is extracted from the aqueous layer into the ethyl acetate in this process.
Progress of the extraction of methyl red (the colored compound) from the acidic aqueous layer (bottom) into the organic layer (top). The inversions were done slowly in order to see the extraction stepwise. With even gentle mixing, the methyl red extracts rapidly.


Prepare the Setup (for single extraction)
1. Obtain a separatory funnel (Fig.4 a).- a) If the separatory funnel has a Teflon stopcock, reassemble the stopcock if it was taken apart to dry, placing the parts in the appropriate order (Fig.4 b). Be sure that the Teflon stopcock is moderately tight so that it can still easily turn, but is not so loose that liquid can seep around the joint.
- b) If using a glass stopcock (Fig.4 c), it likely needs no further preparation. There should be a very thin layer of grease used to seal the stopcock and prevent freezing. If both glass and Teflon stopcocks are available, Teflon is a better choice, as there is always a possibility that solvent can dissolve the grease used with glass stopcocks and contaminate the sample.
- c) Also obtain a stopper (Teflon or ground glass) that fits well in the top joint of the funnel (Fig.4 a).
2. Place the separatory funnel in a ring clamp attached to a ring stand or latticework. The funnels are easy to break, so cushion the funnel in the metal clamp using pieces of slit rubber or plastic tubing (Fig.4 d).
Add the Solutions (for single extraction)
3. Before pouring anything into a separatory funnel, be sure that the stopcock is in the "closed" position, where the stopcock is horizontal (Fig.5 a). As a fail-safe, always position an Erlenmeyer flask beneath the separatory funnel before pouring (Fig.5 b). This can catch liquid in case the stopcock is accidentally left open, or if the stopcock is loose and liquid leaks through unintentionally.
4. Using a funnel, pour the liquid to be extracted into the separatory funnel (Fig.5 b). A separatory funnel should never be used with a hot or warm liquid. The ground glass joint atop a separatory funnel is more prone to stick to the stopper if there was liquid in the joint at some point. Pouring liquid into the separatory funnel using a short-stemmed funnel avoids getting the joint wet, so that it will be less likely to freeze during mixing.
3. Before pouring anything into a separatory funnel, be sure that the stopcock is in the "closed" position, where the stopcock is horizontal (Fig.5 a). As a fail-safe, always position an Erlenmeyer flask beneath the separatory funnel before pouring (Fig.5 b). This can catch liquid in case the stopcock is accidentally left open, or if the stopcock is loose and liquid leaks through unintentionally.
4. Using a funnel, pour the liquid to be extracted into the separatory funnel (Fig.5 b). A separatory funnel should never be used with a hot or warm liquid. The ground glass joint atop a separatory funnel is more prone to stick to the stopper if there was liquid in the joint at some point. Pouring liquid into the separatory funnel using a short-stemmed funnel avoids getting the joint wet, so that it will be less likely to freeze during mixing.
Pour a quantity of the extractive solvent into the separatory funnel, as indicated by the procedure (Fig.5 c). It is unnecessary to use precise quantities of solvent for extractions, and the volumes can be measured in a graduated cylinder. If a procedure calls for 20mL of solvent, it is acceptable if between 20-25ml is used each time.
Mix the Solutions (for single extraction)
6. Place the stopper on the funnel, and hold the funnel such that the fingers of one hand securely cover the stopper, while the other hand grips the bottom of the funnel (Fig.6 a).
7. Gently invert the funnel (Fig.6 b), and swirl the mixture a little. Although it is not uncommon for some liquid to creep into the ground glass joint when inverted, it should be minimal. If liquid drips onto your fingers or gloves when you invert the funnel, the stopper is probably the wrong size.
Mix the Solutions (for single extraction)
6. Place the stopper on the funnel, and hold the funnel such that the fingers of one hand securely cover the stopper, while the other hand grips the bottom of the funnel (Fig.6 a).
7. Gently invert the funnel (Fig.6 b), and swirl the mixture a little. Although it is not uncommon for some liquid to creep into the ground glass joint when inverted, it should be minimal. If liquid drips onto your fingers or gloves when you invert the funnel, the stopper is probably the wrong size.
8. Pressure may build up inside the separatory funnel when solutions are mixed, so immediately after swirling, and with the funnel still inverted, "vent" the funnel by briefly opening the stopcock to allow for a release of pressure (Fig.6 c). Pressure builds in the funnel as solvent evaporates into the headspace and contributes additional vapor to the initial ∼1 atmosphere of air pressure in the funnel. With highly volatile solvents (like diethyl ether), a definite "swoosh" can be heard upon venting, and small amounts of liquid may even sputter out the stopcock. If liquid spits out the stopcock, try to allow it to drain back into the funnel. The noise associated with venting normally ceases after the second or third inversions, as the headspace becomes saturated with solvent vapors and the pressures inside and outside the funnel are equalized.
Safety note: Never point the stopcock toward someone as you vent, as it's possible some liquid may splatter onto him or her.
9. Close the stopcock and mix the solutions a bit more vigorously, periodically stopping to vent the system. There are differences of opinion on how vigorously solutions should be mixed in separatory funnels, and for how long. As a general guide, a mild mixing for 10-20 seconds should be enough. With some solutions (e.g. dichloromethane), care should be taken to not shake too vigorously, as these solutions often form emulsions (where the interface between the solutions doesn't clarify). With solutions prone to emulsions, a funnel should be gently rocked for one minute.
10. Place the separatory funnel upright in the ring clamp to allow the layers to fully separate. The interface between the layers should settle rather quickly, often within 10 seconds or so. If the interface is clouded or not well-defined (an emulsion has formed), see the troubleshooting section for tips.
Separate the Layers (for single extraction)
11. Liquid will not drain well from a separatory funnel if the stopper remains on, as air cannot enter the funnel to replace the displaced liquid. If liquid did drain from the funnel without replacement by an equal volume of air, a negative pressure would form in the funnel. Thus, before draining liquid from a separatory funnel, remove the stopper (Fig.7 a).
12. Drain the majority of the bottom layer into a clean Erlenmeyer flask, positioning the ring clamp so that the tip of the separatory funnel is nestled in the Erlenmeyer flask to prevent splashing (Fig.7 b). Stop draining when the interface is within 1 cm of the bottom of the stopcock.
13. Gently swirl the funnel to dislodge any droplets clinging to the glass (Fig.7 c). A glass stirring rod can be used to knock down stubborn clinging droplets.
14. Further, drain the bottom layer, stopping when the interface just enters the stopcock chamber (Fig.7 d). Label the Erlenmeyer flask (e.g. "bottom layer").
Safety note: Never point the stopcock toward someone as you vent, as it's possible some liquid may splatter onto him or her.
9. Close the stopcock and mix the solutions a bit more vigorously, periodically stopping to vent the system. There are differences of opinion on how vigorously solutions should be mixed in separatory funnels, and for how long. As a general guide, a mild mixing for 10-20 seconds should be enough. With some solutions (e.g. dichloromethane), care should be taken to not shake too vigorously, as these solutions often form emulsions (where the interface between the solutions doesn't clarify). With solutions prone to emulsions, a funnel should be gently rocked for one minute.
10. Place the separatory funnel upright in the ring clamp to allow the layers to fully separate. The interface between the layers should settle rather quickly, often within 10 seconds or so. If the interface is clouded or not well-defined (an emulsion has formed), see the troubleshooting section for tips.
Separate the Layers (for single extraction)
11. Liquid will not drain well from a separatory funnel if the stopper remains on, as air cannot enter the funnel to replace the displaced liquid. If liquid did drain from the funnel without replacement by an equal volume of air, a negative pressure would form in the funnel. Thus, before draining liquid from a separatory funnel, remove the stopper (Fig.7 a).
12. Drain the majority of the bottom layer into a clean Erlenmeyer flask, positioning the ring clamp so that the tip of the separatory funnel is nestled in the Erlenmeyer flask to prevent splashing (Fig.7 b). Stop draining when the interface is within 1 cm of the bottom of the stopcock.
13. Gently swirl the funnel to dislodge any droplets clinging to the glass (Fig.7 c). A glass stirring rod can be used to knock down stubborn clinging droplets.
14. Further, drain the bottom layer, stopping when the interface just enters the stopcock chamber (Fig.7 d). Label the Erlenmeyer flask (e.g. "bottom layer").
15. Pour out the top layer from the top of the separatory funnel into another clean Erlenmeyer flask (Fig.8 a), making sure to again label this flask (Fig.8 b). It is proper technique to drain the bottom layer through the stopcock, and to pour out the top layer from the top of the funnel. This method minimizes re-mixing the solutions, as only the lower layer touches the stem of the funnel.
16. Never throw away any liquids from an extraction until you are absolutely sure that you have the desired compound. Undesired layers can be properly disposed of when the desired compound is in your hands (e.g. after the rotary evaporator has removed the solvent).
Mistakes made during extractions (e.g. carrying on with the wrong layer), can be solved as long as the solutions have not been placed in the waste container! The layers should also be saved until after evaporation because the desired compound may not be very soluble in the solvent used. If the compound failed to extract in one solvent, a different solvent could be tried later, again only if the layers had not yet been thrown away.
Clean Up (for single extraction)
To clean a separatory funnel, first rinse it with acetone into a waste container. Then wash the funnel with soap and water at your bench top. Disassemble the Teflon stopcock (if used). After rinsing with distilled water, allow the parts to dry separated in your locker (Fig.8 c).
Mistakes made during extractions (e.g. carrying on with the wrong layer), can be solved as long as the solutions have not been placed in the waste container! The layers should also be saved until after evaporation because the desired compound may not be very soluble in the solvent used. If the compound failed to extract in one solvent, a different solvent could be tried later, again only if the layers had not yet been thrown away.
Clean Up (for single extraction)
To clean a separatory funnel, first rinse it with acetone into a waste container. Then wash the funnel with soap and water at your bench top. Disassemble the Teflon stopcock (if used). After rinsing with distilled water, allow the parts to dry separated in your locker (Fig.8 c).
Multiple Extractions.
In this section are stepwise instructions on how to extract an aqueous solution with an organic solvent that is less dense than water (the organic layer will be on the top). As an example, the instructions are written to extract an aqueous solution three times using 25 ml diethyl ether each time (3×25 ml diethyl ether). A procedural summary of the first two extractions is in Fig.9 Two extractions when the organic layer is on the top.
Extraction #1
1. Perform a single extraction using approximately 25 ml of diethyl ether (an exact amount is not necessary), as described previously, making sure to appropriately label each layer (e.g. "top organic layer" and "bottom aqueous layer").
Extraction #2
2. Return the aqueous layer to the separatory funnel. There is no need to wash the funnel in between extractions.
3. Add a fresh 25 ml portion of diethyl ether to the separatory funnel. Stopper the funnel, invert and shake with venting, then allow the layers to separate.
At this step, there should be two layers in the separatory funnel. If two layers aren't present, it's likely that the wrong layer was added to the funnel in step 2 (a common mistake). One way to test if this was the mistake is to add a bit of water from a squirt bottle. If the layer returned to the separatory funnel is the organic layer (incorrect), the squirt bottle water will not mix with the solution, and will instead fall as droplets to the bottom. If the organic layer (incorrect) was accidentally returned to the separatory funnel, there is no harm done, as the organic layer was simply diluted. Pour the liquid back into the flask designed for the organic layer, and instead add the aqueous solution to the funnel.
4. Drain the bottom aqueous layer into an Erlenmeyer flask: it is acceptable to use the same flask that was used for the aqueous layer in the first extraction (that may have been labeled "bottom aqueous layer").
5. Since it is most common to combine the organic layers in multiple extractions, the top organic layer can be poured out of the separatory funnel into the same flask that was used for the organic layer in the first extraction (that may have been labeled "top organic layer"). In this flask, there should be roughly 50 ml of diethyl ether from the two extractions.
Extraction #3
6. Repeat the extraction a third time by adding the aqueous layer from the second extraction into the separatory funnel, followed by another fresh 25 ml portion of diethyl ether. Stopper the funnel, invert and shake with venting, then allow the layers to separate.
7. Drain the aqueous layer into the appropriate flask, and again pour the top layer into the organic layer flask, where there should be roughly 75 ml of diethyl ether from the three extractions.
1. Perform a single extraction using approximately 25 ml of diethyl ether (an exact amount is not necessary), as described previously, making sure to appropriately label each layer (e.g. "top organic layer" and "bottom aqueous layer").
Extraction #2
2. Return the aqueous layer to the separatory funnel. There is no need to wash the funnel in between extractions.
3. Add a fresh 25 ml portion of diethyl ether to the separatory funnel. Stopper the funnel, invert and shake with venting, then allow the layers to separate.
At this step, there should be two layers in the separatory funnel. If two layers aren't present, it's likely that the wrong layer was added to the funnel in step 2 (a common mistake). One way to test if this was the mistake is to add a bit of water from a squirt bottle. If the layer returned to the separatory funnel is the organic layer (incorrect), the squirt bottle water will not mix with the solution, and will instead fall as droplets to the bottom. If the organic layer (incorrect) was accidentally returned to the separatory funnel, there is no harm done, as the organic layer was simply diluted. Pour the liquid back into the flask designed for the organic layer, and instead add the aqueous solution to the funnel.
4. Drain the bottom aqueous layer into an Erlenmeyer flask: it is acceptable to use the same flask that was used for the aqueous layer in the first extraction (that may have been labeled "bottom aqueous layer").
5. Since it is most common to combine the organic layers in multiple extractions, the top organic layer can be poured out of the separatory funnel into the same flask that was used for the organic layer in the first extraction (that may have been labeled "top organic layer"). In this flask, there should be roughly 50 ml of diethyl ether from the two extractions.
Extraction #3
6. Repeat the extraction a third time by adding the aqueous layer from the second extraction into the separatory funnel, followed by another fresh 25 ml portion of diethyl ether. Stopper the funnel, invert and shake with venting, then allow the layers to separate.
7. Drain the aqueous layer into the appropriate flask, and again pour the top layer into the organic layer flask, where there should be roughly 75 ml of diethyl ether from the three extractions.
Troubleshooting.
This section descries common problems and solutions in extractions.There is Only One Layer
The most common reason for having only one layer in a separatory funnel when there should be two (as in when the procedure tells you to "separate the layers"), is to have made a mistake. What likely happened is that the wrong layer was added to the separatory funnel - for example, the organic layer was unknowingly added instead of the aqueous layer. When organic solvent is added to an organic layer in the separatory funnel, the result is only one layer. The mistake can be remedied as long as the layers have not yet been thrown away! If the correct layer is added to the funnel, everything will work out as planned. To prevent making this mistake in the future, be sure to label the Erlenmeyer flasks. Also, be sure to never throw away a layer until you are absolutely sure that you've done everything correctly.
An occasional reason that only one layer forms in a separatory funnel is if there are large quantities of compounds present that dissolve in both solvents, for example if large amounts of ethanol are present, which dissolve well in both aqueous and organic solvents. In this situation, the best approach is to remove the troublesome compound (i.e. the ethanol) on a rotary evaporator before extraction.
There are Three Layers
The most common reason for three layers in a separatory funnel is inadequate mixing (Fig.10 a). If the funnel is shaken with more vigor, it will likely settle into two layers (Fig.10 b). It is also possible that a middle third layer is an emulsion, where the two layers are not fully separated.
The most common reason for having only one layer in a separatory funnel when there should be two (as in when the procedure tells you to "separate the layers"), is to have made a mistake. What likely happened is that the wrong layer was added to the separatory funnel - for example, the organic layer was unknowingly added instead of the aqueous layer. When organic solvent is added to an organic layer in the separatory funnel, the result is only one layer. The mistake can be remedied as long as the layers have not yet been thrown away! If the correct layer is added to the funnel, everything will work out as planned. To prevent making this mistake in the future, be sure to label the Erlenmeyer flasks. Also, be sure to never throw away a layer until you are absolutely sure that you've done everything correctly.
An occasional reason that only one layer forms in a separatory funnel is if there are large quantities of compounds present that dissolve in both solvents, for example if large amounts of ethanol are present, which dissolve well in both aqueous and organic solvents. In this situation, the best approach is to remove the troublesome compound (i.e. the ethanol) on a rotary evaporator before extraction.
There are Three Layers
The most common reason for three layers in a separatory funnel is inadequate mixing (Fig.10 a). If the funnel is shaken with more vigor, it will likely settle into two layers (Fig.10 b). It is also possible that a middle third layer is an emulsion, where the two layers are not fully separated.
There is Insoluble Material at the Interface.
A small amount of insoluble film between two layers is not uncommon during an extraction. Polymeric materials tend to rest between layers as solvent interactions are minimized at the interface. A minor film is not something to worry about because if a small amount does make it into the organic layer, a subsequent drying and filtration step will often remove it.
The Interface Cannot be Seen.
On occasion, the compounds in a separatory funnel are so dark that they obscure the interface between the two layers. If this happens, there are several methods that might help you see the interface. One is to hold the separatory funnel up to the light, or to shine a flashlight onto the glass (Fig.11 b). Additional light sometimes allows you to see the interface. A second method is to carefully observe the layers while tilting the funnel back and forth to the side (Fig.11 c). Your eye can sometimes pick up on subtle differences in the way the liquids flow. A third method is to add a bit more solvent to the funnel to somewhat dilute one of the layers, or to add a different solvent to alter the index of refraction.
The Layers Don't Separate Well (An Emulsion Formed)
Emulsions can happen for several reasons:
1. The density of each layer may be so similar that there is weak motivation for the liquids to separate.2. There may be soap-like compounds or other emulsifying agents present that dissolve some components in one another.
Emulsions can be very difficult to rectify, and it's best if they are avoided in the first place by shaking solutions that are prone to emulsions (e.g. dichloromethane with highly basic or dense solutions) gently in the separatory funnel. Nonetheless, if an emulsion does form, there are some ways to attempt to clarify them:
- a) For mild emulsions, gently swirl the layers and try to knock down suspended droplets with a glass stirring rod.
- b) Allow the solution to sit for a period of time (even until the next lab period) if possible. With enough time, some solutions do settle out on their own. This of course may not be practical.
- c) For small volumes, use a centrifuge if one is available. A centrifuge hastens the process of letting an emulsion settle on its own. Remember that a centrifuge needs to be balanced, or it may wobble off the bench top. Divide the solutions equally, putting tubes of equal volume opposite one another inside the centrifuge.
- d) If an emulsion is formed because the two layers have similar densities, try to alter the density of each layer to make them more different. To help clarify an emulsion, try to decrease the density of the top layer or increase the density of the bottom layer. For example, if an emulsion occurs with ethyl acetate (top layer) and an aqueous solution (bottom layer), add some NaCl. NaCl will dissolve in the aqueous layer and increase the density of the aqueous solution. Alternatively, add additional ethyl acetate, which will dilute the organic layer and lower its density. As a last resort, add some pentane, which will mix with the top organic layer and decrease its density (pentane is one of the least dense organic solvents). The addition of pentane is used as a final effort, as it will negatively affect the ability of the organic layer to extract somewhat polar compounds.
If an emulsion occurs with an aqueous solution (top layer) and dichloromethane (bottom layer), add some water from a squirt bottle to dilute the top layer and decrease its density. This method worked well to clarify the emulsion in Fig 13 c, as evidenced by Fig 13 d. - c) Try decreasing the solubility of one component in the other. One method is to add NaCl or NH4Cl to the separatory funnel, which dissolves in the aqueous layer and decreases the ability of organic compounds to dissolve in water ("salting out").
Acid-Base Extraction
How They Work.
A modification of the extractions previously discussed in this chapter is to perform a chemical reaction in the separatory funnel in order to change the polarity and therefore partitioning of a compound in the aqueous and organic layers. A common method is to perform an acid-base reaction, which can convert some compounds from neutral to ionic forms (or vice versa).
For example, imagine that a mixture of benzoic acid and cyclohexane is dissolved in an organic solvent like ethyl acetate in a separatory funnel. To separate the components, a water wash may be attempted to remove benzoic acid, but benzoic acid is not particularly water-soluble due to its nonpolar aromatic ring, and only small amounts would be extracted into the aqueous layer (Fig.14 a).
For example, imagine that a mixture of benzoic acid and cyclohexane is dissolved in an organic solvent like ethyl acetate in a separatory funnel. To separate the components, a water wash may be attempted to remove benzoic acid, but benzoic acid is not particularly water-soluble due to its nonpolar aromatic ring, and only small amounts would be extracted into the aqueous layer (Fig.14 a).
Separation of a mixture of benzoic acid and cyclohexane is however possible using a wash with a base, such as NaOH. Due to its acidic nature, benzoic acid can undergo a reaction with NaOH as follows, resulting in the carboxylate salt sodium benzoate.
The solubility properties of carboxylic acids are substantially different from their corresponding carboxylate salts. Sodium salicylate is roughly 350 times more soluble in water than salicylic acid due to its ionic character (Fig.15), and it is rather insoluble in organic solvents such as diethyl ether.
Therefore, a wash with NaOH would convert benzoic acid into its ionic carboxylate form, which would then be more soluble in the aqueous layer, allowing for the sodium benzoate to be extracted into the aqueous layer. Cyclohexane would remain in the organic layer as it has no affinity for the aqueous phase, nor can react with NaOH in any way. In this manner, a mixture of benzoic acid and cyclohexane can be separated (Fig.14 b). The aqueous layer may be later acidified with HCl(aq) if desired to convert the benzoic acid back to its neutral form.
Sodium Bicarbonate Washes
An acid-base extraction can be used to extract carboxylic acids from the organic layer into the aqueous layer. As was discussed in the previous section, NaOH can be used to convert a carboxylic acid into its more water-soluble ionic carboxylate form. However, if the mixture contains a desired compound that can react with NaOH, a milder base such as sodium bicarbonate should be used. A similar reaction occurs:
Sodium Bicarbonate Washes
An acid-base extraction can be used to extract carboxylic acids from the organic layer into the aqueous layer. As was discussed in the previous section, NaOH can be used to convert a carboxylic acid into its more water-soluble ionic carboxylate form. However, if the mixture contains a desired compound that can react with NaOH, a milder base such as sodium bicarbonate should be used. A similar reaction occurs:
One difference in using the base NaHCO3 instead of NaOH is that the byproduct carbonic acid (H2CO3) can decompose to water and carbon dioxide gas. When shaking an acidic solution with sodium bicarbonate in a separatory funnel, care should be taken to swirl gently and vent more frequently to release pressure from the gas.
An example of a reaction that often uses sodium bicarbonate wash in the work-up is a Fischer Esterification reaction. To demonstrate, benzoic acid was refluxed in ethanol along with concentrated sulfuric acid in order to form ethyl benzoate (Fig.16 a and b). A TLC plate of the reaction mixture at 1 hour of reflux showed residual unreacted carboxylic acid (Fig.16 c), which is not uncommon due to the energetics of the reaction.
An example of a reaction that often uses sodium bicarbonate wash in the work-up is a Fischer Esterification reaction. To demonstrate, benzoic acid was refluxed in ethanol along with concentrated sulfuric acid in order to form ethyl benzoate (Fig.16 a and b). A TLC plate of the reaction mixture at 1 hour of reflux showed residual unreacted carboxylic acid (Fig.16 c), which is not uncommon due to the energetics of the reaction.
The residual carboxylic acid can be removed from the desired ester product using an acid-base extraction in a separatory funnel. A wash with sodium bicarbonate converts benzoic acid into its more water-soluble sodium benzoate form, extracting it into the aqueous layer (Fig. 17). Additionally, the sodium bicarbonate neutralizes the catalytic acid in this reaction.
Sodium bicarbonate is preferable to NaOH in this process, as it is a much weaker base; washing with NaOH could cause hydrolysis of the ester product.
Mixtures of Acids and Bases
As has been discussed previously, the acid-base properties of compounds can be utilized to selectively extract certain compounds from mixtures. This strategy can be extended to other examplesMixtures of Acids and Bases
Extracting Bases
Basic compounds such as amines can be extracted from organic solutions by shaking them with acidic solutions to convert them into more water-soluble salts. In this way, they can be extracted from an organic layer into an aqueous layer.Extracting Carboxylic Acids vs. Phenols
As previously discussed, carboxylic acids can be extracted from an organic layer into an aqueous layer by shaking them with basic solutions, which converts them into their more water-soluble salts.
As previously discussed, carboxylic acids can be extracted from an organic layer into an aqueous layer by shaking them with basic solutions, which converts them into their more water-soluble salts.
A similar reaction occurs with phenols (PhOH), and they too can be extracted into an aqueous NaOH layer (Fig.18 a).
However, phenols are considerably less acidic than carboxylic acids, and are not acidic enough to react completely with NaHCO3, a weaker base. Therefore, a solution of bicarbonate can be used to separate mixtures of phenols and carboxylic acids (Fig.18 b).
However, phenols are considerably less acidic than carboxylic acids, and are not acidic enough to react completely with NaHCO3, a weaker base. Therefore, a solution of bicarbonate can be used to separate mixtures of phenols and carboxylic acids (Fig.18 b).
Extracting Acid, Base, and Neutral Compounds
The acid-base properties previously discussed allow for a mixture containing acidic (e.g. RCO2H), basic (e.g. RNH2), and neutral components to be purified through a series of extractions, as summarized in Fig.19 (which uses an organic solvent less dense than water).
The acid-base properties previously discussed allow for a mixture containing acidic (e.g. RCO2H), basic (e.g. RNH2), and neutral components to be purified through a series of extractions, as summarized in Fig.19 (which uses an organic solvent less dense than water).
It is assumed that readers conducting this type of experiment are familiar with performing single and multiple extractions. In this section are described differences between general extraction procedures and the process as summarized in Fig. 19.
1. Isolating the Acidic component:
a) When the acidic component is in the aqueous layer in an Erlenmeyer flask, it can be converted back to the neutral component through addition of 2M HCl(aq) until the solution gives a pH of 3-4 (as determined by pH paper). If large quantities of acid are present such that acidification would require too great a volume of 2M HCl(aq), concentrated HCl(aq) may be instead added dropwise. Lower concentrations of Hcl(aq) are less hazardous, but increasing the volume of the aqueous layer by a large amount would affect the efficiency of subsequent extractions and filtering steps.
b) After acidification, two routs may be taken, depending on if the acidic component is solid or liquid.
1. Isolating the Acidic component:
a) When the acidic component is in the aqueous layer in an Erlenmeyer flask, it can be converted back to the neutral component through addition of 2M HCl(aq) until the solution gives a pH of 3-4 (as determined by pH paper). If large quantities of acid are present such that acidification would require too great a volume of 2M HCl(aq), concentrated HCl(aq) may be instead added dropwise. Lower concentrations of Hcl(aq) are less hazardous, but increasing the volume of the aqueous layer by a large amount would affect the efficiency of subsequent extractions and filtering steps.
b) After acidification, two routs may be taken, depending on if the acidic component is solid or liquid.
- If a solid forms upon acidification of the ionic salt, it can be collected through suction filtration. This method should only be used if large quantities of large-sized crystals are seen. If fine crystals form (which are quite common), they will clog the filter paper and interfere with adequate drainage. If only a small amount of solid is seen compared to the theoretical quantity, it is likely the compound is quite water-soluble, and filtration would lead to low recovery.
- If no solid forms upon acidification (or if fine crystals or low quantity of solid forms), extract the acidic component back into an organic solvent (×3). As a general rule of thumb, use one-third as much solvent for the extractions as the original layer (e.g. if using 100ml aqueous solution, extract with 33ml organic solvent each time). Be sure to first cool the aqueous solution in an ice bath before extraction if the acidification created noticeable heat. Follow up with a brine wash (×1) if using diethyl ether or ethyl acetate, dry with a drying agent, and remove the solvent via rotary evaporator to leave the pure acidic component.
2. Isolating the Basic component:
Use a similar process as the isolation of the acidic component, except basify the solution using 2M NaOH(aq) until it gives a pH of 9-10 as determined by pH paper.
3. Isolating the Neutral component:
The neutral component will be the "leftover" compound in the organic layer. To isolate, wash with brine (×1) if using diethyl ether or ethyl acetate, dry with a drying agent, and remove the solvent via rotary evaporator to leave the pure neutral component.
Use a similar process as the isolation of the acidic component, except basify the solution using 2M NaOH(aq) until it gives a pH of 9-10 as determined by pH paper.
3. Isolating the Neutral component:
The neutral component will be the "leftover" compound in the organic layer. To isolate, wash with brine (×1) if using diethyl ether or ethyl acetate, dry with a drying agent, and remove the solvent via rotary evaporator to leave the pure neutral component.
Conclusion.
Extraction methods are using for extracting some substances from mixture. These substances may be base, acid or neutral (polar or non-polar). For instance, this method is use during amphetamine production in stage of Decantation: collect the top layer containing amphetamine base in alcohol. It can be dried a little with anhydrous magnesium sulfate, and the slag can be additionally extracted with a non-polar solvent (ether, benzene, toluene), the solvent is then evaporated. Mephedron production include manipulation with separating funnel and extraction. Also, the Acid-base extraction is used in the purification of some psychoactive substances from impurities.
Attachments
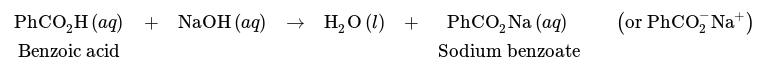
Last edited: